The Edit Menu
This menu contains most of the functions that change the entries. Note that the changes will not be saved back to the original files until one of the save functions in the File menu is used (see the section called Save An Entry).
Undo
This function will undo the last change that was made using the Edit or Create menus. Up to 20 changes can be undone. This menu item is only enabled when there is something to undo. This limit can be changed in the options file (see the section called undo_levels in Chapter 5). [shortcut key: U]
Redo
This function will redo the last undo operation.
Selected Features in Editor
Open an edit window for each selected feature. [shortcut key: E]
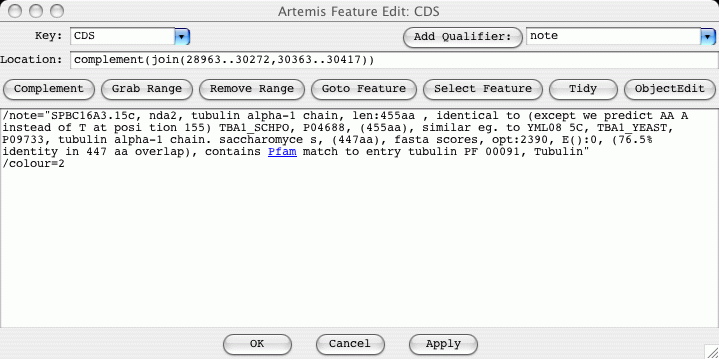
From the top down the edit window has these parts:
At the top left is a selector for choosing the key of the feature. This only contains a subset of the legal keys. The subset can be changed by changing the common_keys option in the options file (see the section called common_keys in Chapter 5).
At the top right of the edit window is a selector for adding a qualifier. For example choosing note from the menu will insert /note="" into the qualifier edit area.
Just below the key and qualifier selector is the location entry field. Artemis understands most of the EMBL location syntax, including joins, complements, ranges with non-exact ends (eg. (100.200)..>350) and references to other entries (eg. join(100..200,SPB23A1:100..200)).
Below the location is a row of buttons:
The Complement button will complement the contents of the location field.
The Grab Range button will grab the currently selected range into the location field.
The Remove Range button will remove the selected bases from the location string. This is normally used to create an intron in a feature.
Pressing the Goto Feature button has the same effect as the "Start of Selection" item in the "Goto" menu. (See the section called Start of Selection for more).
The Select Feature button selects this feature (in the same way as clicking on the feature in one of the views).
The centre of the edit window contains the qualifier entry section. The qualifiers should be entered the in same way the appear in the feature table part of an EMBL entry, but without the leading FT and spaces.
The bottom of the window contains three buttons. The OK button will update the feature with the changes that have been made by the user and will then close the edit window. The Cancel button will discard the changes and then close the window. The Apply will make the changes, but will not close the window. Before any changes are made the location and the qualifiers are checked for formatting errors. Any errors will brought to the attention of the user through the use of annoying pop-up boxes. No changes will be performed until there all errors have been fixed.
Subsequence (and Features)
Make a copy (in a new edit window) of the selected bases and the features in that range. Any features that overlap the end of the range will be truncated.
Qualifier(s) of Selected Feature
Change ...
This function allows the user to add or change qualifiers on all the selected features in one operation. The main part of the window acts like the qualifier editing field of the feature edit window (see the section called Selected Features in Editor).
Remove ...
This function allows the user to remove all qualifiers with a particular name from all the selected features.
Convert ...
This function allows the user to convert all qualifiers of a particular type to another qualifier for all the selected features.
Selected Feature(s)
Duplicate
Make a copy of each selected feature. Each new feature will be added just after the original in the same entry as the original. [shortcut key: D]
Merge
Create a new feature that contains all the exons and qualifiers of the selected features. The selected features must all have the same key. [shortcut key: M]
Unmerge
If the selection contains exactly two exons and those exons are adjacent in the same feature, split the feature into two pieces between the exons. The original feature is truncated and a new feature is created. The qualifiers of the old feature are copied to new feature.
Unmerge All Segments
All exons in a feature are unmerged.
Delete
Remove each selected feature from it's entry. [shortcut key: control-delete]
Delete Exons
Delete the selected exons. The last exon of a feature can't be deleted (delete the whole feature instead).
Remove Introns
Delete the selected introns.
Move Selected Features To
Move the selected features to another entry. Choose the destination entry from the sub-menu.
Copy Selected Features To
Copy the selected features to another entry. Choose the destination entry from the sub-menu.
Trim Selected Features
To Met
For each of the selected features this function will attempt to move the start position to the first ATG in the feature if the feature does not already start on a ATG codon. If there is no ATG in the first thirty percent of the bases of the feature the start position will be unchanged. The search will stop at the end of the first segment of a multi-segment feature.
To Any
This works in the same way as "Trim Selected Features To Met", but will attempt to move the start position of the feature to the first TTG, ATG or GTG in the feature if it does not already start on one of those codons. As above it will only search the first thirty percent of the feature bases and will only search the first segment of a multi-segment feature.
To Next Met
For each of the selected features this function will attempt to move the start position to the next ATG in the feature (the first codon is skipped). If there is no ATG in the first thirty percent of the bases of the feature the start position will be unchanged. The search will stop at the end of the first segment of a multi-segment feature. [shortcut key: T]
To Next Any
This works in the same way as "Trim Selected Features To Next Met", but will attempt to move the start position of the feature to the next TTG, ATG or GTG in the feature (the first codon is skipped). As above it will only search the first thirty percent of the feature bases and will only search the first segment of a multi-segment feature. [shortcut key: Y]
Extend Selected Features
To Previous Stop Codon
Extend each of the selected features which do not start on a stop codon so that each feature starts just after the previous stop codon in this reading frame. [shortcut key: Q]
To Next Stop Codon
Extend each of the selected features which do not end on a stop codon so that each feature ends just before the next stop codon in this reading frame.
To Next Stop Codon and Fix
Same as above but in addition this fixes the stop codons.
Fix Stop Codons
Check and fix the stop codons to all the selected features. For each feature if the last codon is a stop codon, then all is well, nothing further is done to the feature. If the last codon is not a stop codon, but the very next codon is a stop codon, then the end of the feature is moved forward by three bases. If both the last codon and the very next codon after the feature are not stop codons, the feature is selected, an error message is displayed and processing stops immediately.
Automatically Create Gene Names
Ask for a gene name prefix (using a text requester), and then give a unique gene name to each CDS feature in the active entries using that prefix. For example if there are four CDS features with locations: "1..500", "complement(100..600)", "200..700" and "complement(300..800)", entering SPBC16A3 will give the four features these names: SPBC16A3.01, SPBC16A3.02c, SPBC16A3.03 and SPBC16A3.04c.
Fix Gene Names
For each selected CDS, add the gene name from the CDS to neighbouring/overlapping mRNA, intron, exon, gene, 5'UTR and 3'UTR features. Warn about inconsistencies such as overlapping CDSs.
Bases
Reverse And Complement
Reverse and complement the sequence and all the features in all the entries (active and inactive).
Reverse And Complement Selected Contig
Reverse and complement the sequence and all the features in a selected contig feature. If this option is used in ACT then all the matches within the contig are also reversed. Any matches extending past the boundary of the contig are deleted. The changes to the comparison file can be saved by right clicking in the comparison window and selecting "Save Comparison File...". However, ideally the comparison between the two sequences should be recalculated.
Delete Selected Bases
Deletes the selected range of bases (if any) from both strands. The deletion will not proceed if the selected range overlaps any features.
Add Bases At Selection
Prompt the user for some bases to insert just before the selected bases. The operation will not proceed if there is no selected range, but bases can be inserted anywhere in the sequence, including inside a feature. The same bases, reversed and complemented, will be inserted at the corresponding place on the opposite strand.
Add Bases From File ...
Prompt the user for the name of a file containing the bases to insert just before the selected bases.
Replace Bases At Selection
Prompt the user for some bases to replace the selected bases.
Contig Reordering ...
Opens a 'Contig Tool' displaying contigs, i.e. with feature keys 'fasta_record' or 'contig'. The former being created automatically for each sequence when a mutiple fasta sequence file is read in. The contigs in this tool can then individually be selected and dragged and dropped to another location. In this way the order of contigs and features within a contig can be changed.
If this is used in ACT then the matches are also reordered with respect to the change in the sequence. If a match spans the boundary of a contig that is being moved then if possible it is split. In some situations where there is a match with 'indels' then this is not possible and the match is deleted. The changes to the comparison file can be saved by right clicking in the comparison window and selecting "Save Comparison File...". However, ideally the comparison between the two sequences should be recalculated.
Header Of Default Entry
Open a edit window containing the header of the default entry. Changes made in the edit window will be applied immediately to the entry provided there are no errors in the formatting of the header.